

Timing Genetic Events
Timing Genetic Events
A revamped technique will help build new biosensors and, one day, record developmental biology's most crucial events.

There’s a new paper out in Nature. I’m writing about it now — in the evening, mere days after my wedding — because it’s a breakthrough in CRISPR-based, genetic recorders. Woe unto me; I cannot control the ceaseless gears of scientific publishing.
Here’s the gist. Cells are messy bags of proteins and RNA. The genome encodes thousands of genes. At any given time, dozens or hundreds of these genes are ‘ON’. That means cells convert sections of DNA to RNA and then into protein. There are experimental techniques, like RNA-seq, that attempt to capture a piece of this process. But RNA-seq is stupid. It says nothing about a gene’s dynamics, and it demands that cells be destroyed. Sure, you could do an RNA-seq experiment at fixed intervals — every 10 minutes or hour — but such an endeavor is strictly reserved for nihilists with no instincts for self-preservation.

So what’s this nonsense about a Nature paper? Well, it describes a biological device, called Retro-Cascorder, that records the timing of genetic events, solely by studying a cell at its endpoint. This paper marks the first time that the specific order of transcriptional events has been recovered from individual cells in this way.
At a high level, the method works by integrating barcoded retrons — DNA sequences common to many bacteria that, after transcription into RNA, are then reverse transcribed into single-stranded DNA — into the genome using CRISPR-Cas integrases. The method is immediately useful for developing living biosensors and, in the future, could record the cellular events that mark an embryo’s miraculous journey into a smiling, speaking, thinking human being.
Order Matters
Seth Shipman is no stranger to storing text, music, or videos in DNA. Back in 2017, Shipman stored the world’s first video — a running horse, photographed by Eadweard Muybridge in 1878 — in the collective genomes of living bacteria. He’s a synthetic DNA storage pioneer, if such a title even exists.

Molecular recorders, then, are nothing new. Biological engineers have long devised elaborate techniques to record cellular processes and permanently store them in DNA. Both recombinase enzymes and CRISPR-Cas gene editing have been used to modify DNA in response to cellular events. DNA sequencing is often used as a ‘readout’ to later study those events. Shipman has also previously used CRISPR-Cas integrases to encode cellular events directly in CRISPR arrays, typically by delivering oligonucleotides into cells.
But no methods have yet managed “to record the temporal order of more than one different biological signal in the CRISPR array of a single cell.” Until now.
The key advancement, here, is that Shipman’s team combined CRISPR-Cas integrases with retrons to “produce customizable DNA in vivo.” Here’s how it works:
Create a ‘library’ of retrons, each of which has a 6-base barcode hidden in its hairpin structure.
Place each custom retron ‘tag’ under the control of a promoter within a cell.
When a gene switches on, the ‘tag’ is transcribed into RNA, and is next reverse transcribed to generate a piece of single-stranded DNA unique to that gene.
The tagged piece of DNA is recognized by a Cas1-Cas2 protein complex, which integrates the DNA into the cell’s CRISPR array. This creates a permanent, genetic record of the transcriptional event. The CRISPR array grows in a fixed direction.
If another gene switches on, its unique tag is also integrated into the CRISPR array. This happens again and again; each retron spacer is added to the CRISPR array.
Sequence the CRISPR array of a single cell. The transcriptional events were recorded linearly, making it easy to retrieve their temporal order.
This device, called Retro-Cascorder, is remarkable for its simplicity. But there are drawbacks. Altering just six bases in each retron sequence changes the relative efficiencies by which those retrons integrate into the CRISPR array. In other words, some ‘tags’ work better than others, and it’s unclear how to scale this approach for hundreds or thousands of genes, if each ‘tag’ varies wildly in efficiency.
Also, the barcodes are only six bases long — the upper limit for this method, then, is only about 4,000 genes. One could make the barcodes longer, but that would presumably diminish the retron efficiencies even further.
Use Case
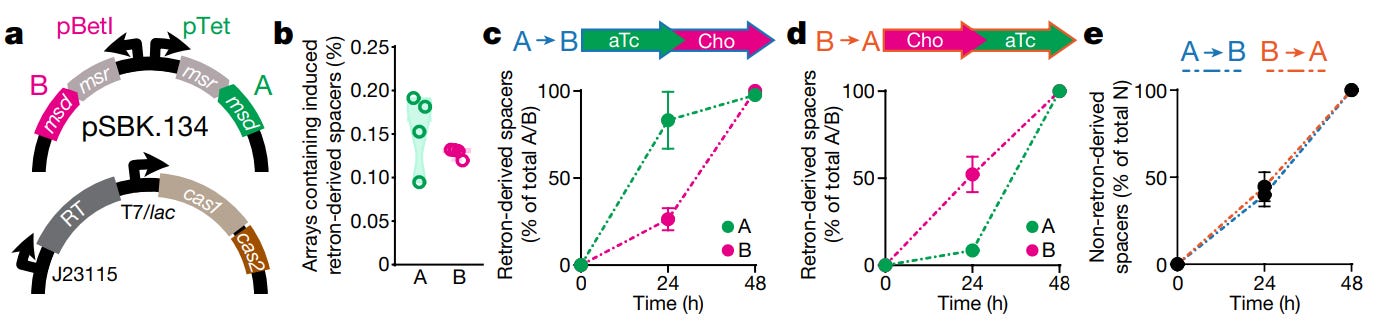
As is often the case, the authors tested their device with a simple experiment. A bacterial cell (E. coli) was endowed with two plasmids, or loops of DNA. One plasmid encodes two separate, non-coding RNAs. Each RNA is tagged with a unique retron barcode (called ‘A’ or ‘B’). Barcode A was placed under the control of a pTet promoter, which switches on when a cell senses the chemical, anhydrotetracycline. Barcode B was placed under the control of a pBetI promoter, which switches on when a cell senses choline chloride. The second plasmid encodes all the other, requisite machinery: Cas1, Cas2, and a reverse transcriptase enzyme.
Now, imagine that you are given a flask of these engineered cells. The cells have been growing for the last 24 hours in liquid containing either anhydrotetracycline or choline chloride. A separate flask holds the other chemical, but no cells.
How would you determine which chemical the cells are bathing in? You’re not a chemist. You’ve never used a mass spectrometer. You are an undergraduate student who possesses an intense familiarity with DNA sequencing.
The obvious experiment is this: Take the flask of cells and centrifuge it. After the cells drop out of solution, extract them. Add them to the flask that holds the second, unknown chemical. Grow them for 24 hours. Extract the cells a second time. Sequence the CRISPR arrays, and the plasmid with the retron barcodes. If the integrated spacers show:
AAAAAAAAAAAAAAAAAAABBBBBBBBBBBBBBBBBBBB
then your answer is clear. Anhydrotetracycline is the chemical in which the cells were first bathed.
This is the experiment that Shipman’s team carried out, and it worked.
Unfortunately, that’s basically where the wet lab experiments end: With a two-event genetic recorder. Instead of building out something more complex — a device that can record 5 or 10 different genes, say — Shipman’s team opted instead to next build a computational model of the Retro-Cascorder. The goal: Understand its theoretical limits.
Each spacer acquisition was modeled as a Poisson process — the average time between acquisitions is known from the experimental data, but the exact timing of events is random — and the team simulated 100 replicates with 1 million arrays each. They tweaked the chemical inducer concentrations, the promoters, the experimental durations, and the number of CRISPR arrays. A key result:
In the range of very short recordings, the system is unable to resolve the order of the signals, but, as the length of the recordings increases, the composite scores converge towards a specific value.
And then…
…even when acquisition rates were decreased, the mean ordering scores across 50 replicates faithfully reflected the order of expression of signals.
The main (theoretical) takeaway, then, is that this system can most faithfully record the timing of genetic events if any of four variables are increased:
(1) signal strength; (2) length of recording; (3) number of reads [DNA sequencing]; and (4) number of replicates. By increasing any of these four parameters, the experimenter can expect greater fidelity of their final temporal recording. When these parameters are decreased, one should expect more noise and variability in the recordings.
Signal strength, presumably, could also be boosted by increasing the efficiency that each retron integrates into the genome.
Prime Time?
My initial reaction to the paper was muted. I want this thing to move into developmental biology labs. But that’s almost certainly a step too far. The tool has only been used in E. coli — based strictly on published experiments; I have no idea what’s going on in Shipman’s lab now, as this paper was submitted way back in June 2021 — and can only record two transcriptional events.
Using Retro-Cascorder to probe the genetic events underlying a cell’s development into a tissue or organ, then, will require a massive technological leap. The device must be able to record dozens or hundreds or thousands of genetic events, across a slew of promoters, with high fidelity and in mammalian cells. Such a momentous hurdle is not stopping Shipman, though. His lab page at the Gladstone Institutes makes that clear:
Using CRISPR, Shipman developed a way to record the order in which genes turn on in a living cell. Now the team is using that technology to understand how the order of gene expression during development drives the formation of different cells [sic] types and tissues.
More soon,
— Niko